High-Harmonic Spectroscopy
High-harmonic spectroscopy is the application of high-harmonic generation to atoms, molecules and the condensed phase to study static and dynamic properties of these systems. High-harmonic spectroscopy offers a number of attractive features that can be exploited to perform unusually accurate and exceptionally sensitive measurements. Our current work focuses on the following topics: Charge migration, laser-induced effects on electronic structure, polar molecules, photochemistry, and electronic wave packets.
A prime advantage of high-harmonic spectroscopy is its temporal resolution. Within each optical cycle of the driving laser pulse, an electron is removed from a molecule, accelerated by the laser field and driven back to recombine with the parent ion. The recombination of an electron emits an attosecond pulse covering a broad range of photon energies (typically several tens of electron-Volts). Each emitted photon energy thereby corresponds to a well-defined time interval between ionization and recombination. Typical time intervals lie between 0.7 and 1.7 fs for 800-nm drivers and scale linearly with the driving wavelength. The amplitude, phase and polarization of these photons carry detailed information about the electronic and geometric structure of the ion at the instant of recombination. This property can be used to extract both structural and electronic dynamics that occurred between ionization and recombination.
Recently, we have developed high-harmonic spectroscopy further to measure and control charge migration in a molecular cation on the attosecond time scale. Figure 1 shows charge migration measured under conditions that mimick the field-free situation.
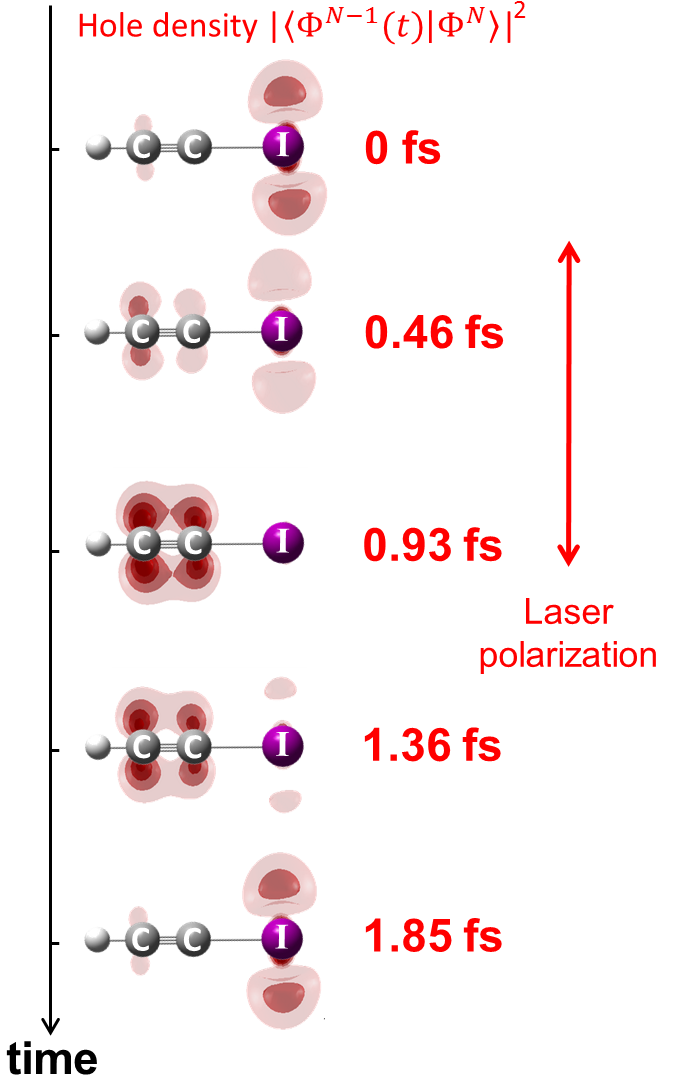
In figure 2 we demonstrate laser control over charge migration, both through the site of electron tunneling and the laser wavelength.
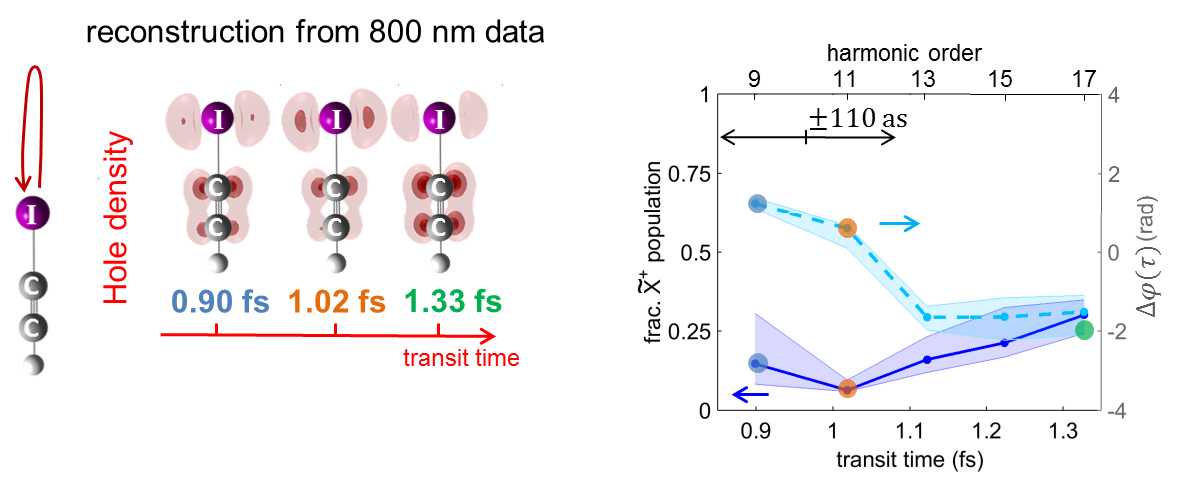
Publications:
[1] P. M. Kraus, B. Mignolet, D. Baykusheva, A. Rupenyan, L. Horny, E. F. Penka, G. Grassi, O. I. Tolstikhin, J. Schneider, F. Jensen, L. B. Madsen, A. D. Bandrauk, F. Remacle, and H. J. Wörner,
"Measurement and laser control of attosecond charge migration in ionized iodoacetylene"
Science 350(6262): 790-795 (2015)
doi: external page 10.1126/science.aab2160
One of the key paradigms in high-harmonic spectroscopy has been to neglect the effect of the laser field on the electronic structure of the studied sample. Whereas this approximation is accurate in rare-gas atoms, it is bound to fail in polarizable molecules.
Using methyl-halide molecules (CH3F and CH3Br) as examples, we have demonstrated the impact of laser-induced modifications to the electronic structure of small molecules [2].
More:
[2] P. M. Kraus, O. I. Tolstikhin, D. Baykusheva, A. Rupenyan, J. Schneider, C. Z. Bisgaard, T. Morishita, F. Jensen, L. B. Madsen, and H. J. Wörner,
Download "Observation of laser-induced electronic structure in oriented polyatomic molecules" (PDF, 763 KB)
Nat. Commun. 6, 7039 (2015)
doi: external page 10.1038/ncomms8039
High-harmonic spectroscopy is especially well suited to study the structure and dynamics of polar molecules because it is naturally sensitive to the inversion symmetry of the generating medium. Spatial orientation of molecules therefore leads to the background-free emission of even harmonics. The relative intensities of the even harmonics encode both the degree of orientation and the electronic asymmetry of the molecule [3]. Whereas impulsive alignment by a single two-color laser pulse only achieves low degrees of orientation, we have demonstrated a two-pulse scheme that achieves macroscopic degrees of orientation (up to 80% of the molecules pointing in the same direction) [4,5].
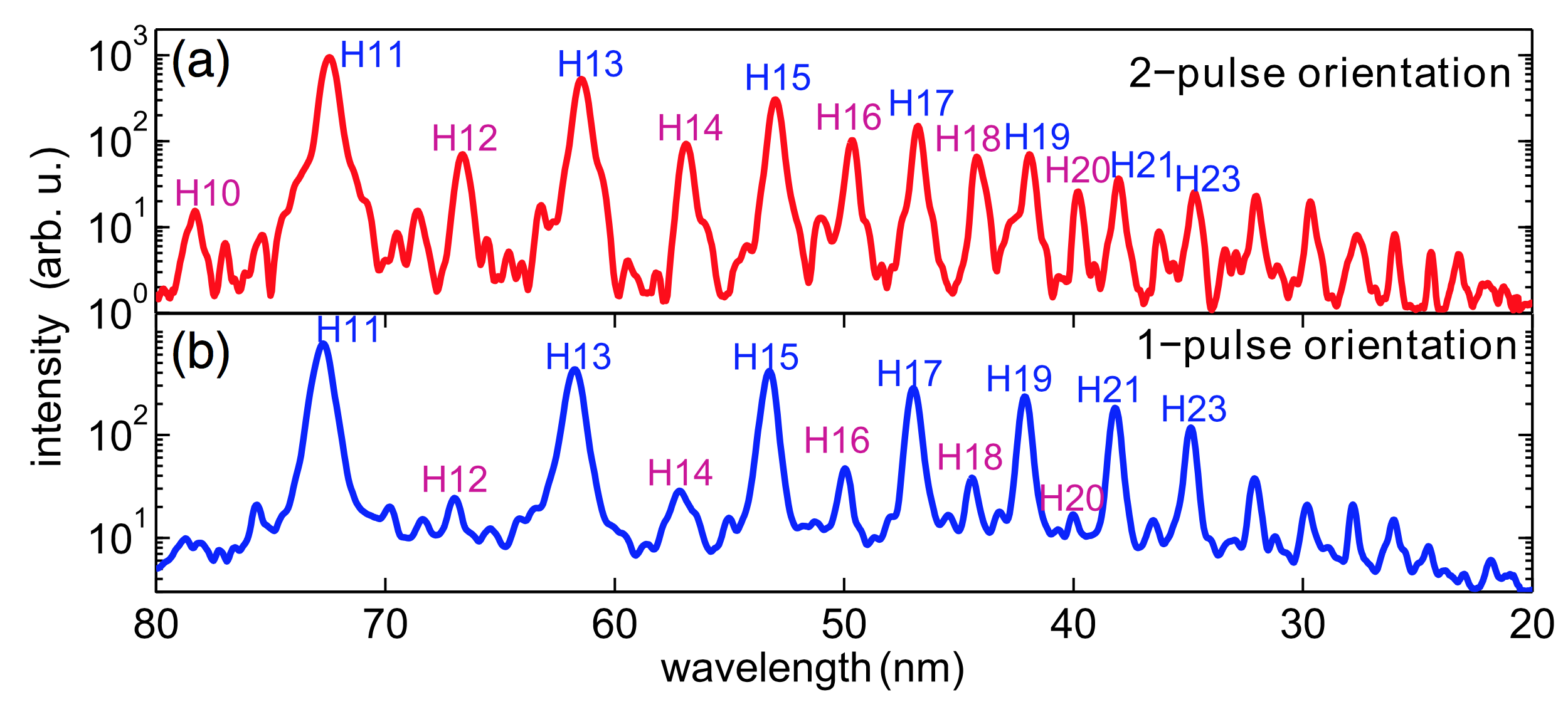
We have moreover shown that a shape resonance in the photoionization continuum of CO leads to a local maximum in the even-to-odd harmonic ratio, revealing the spatial anisotropy of this shape resonance [4,5].
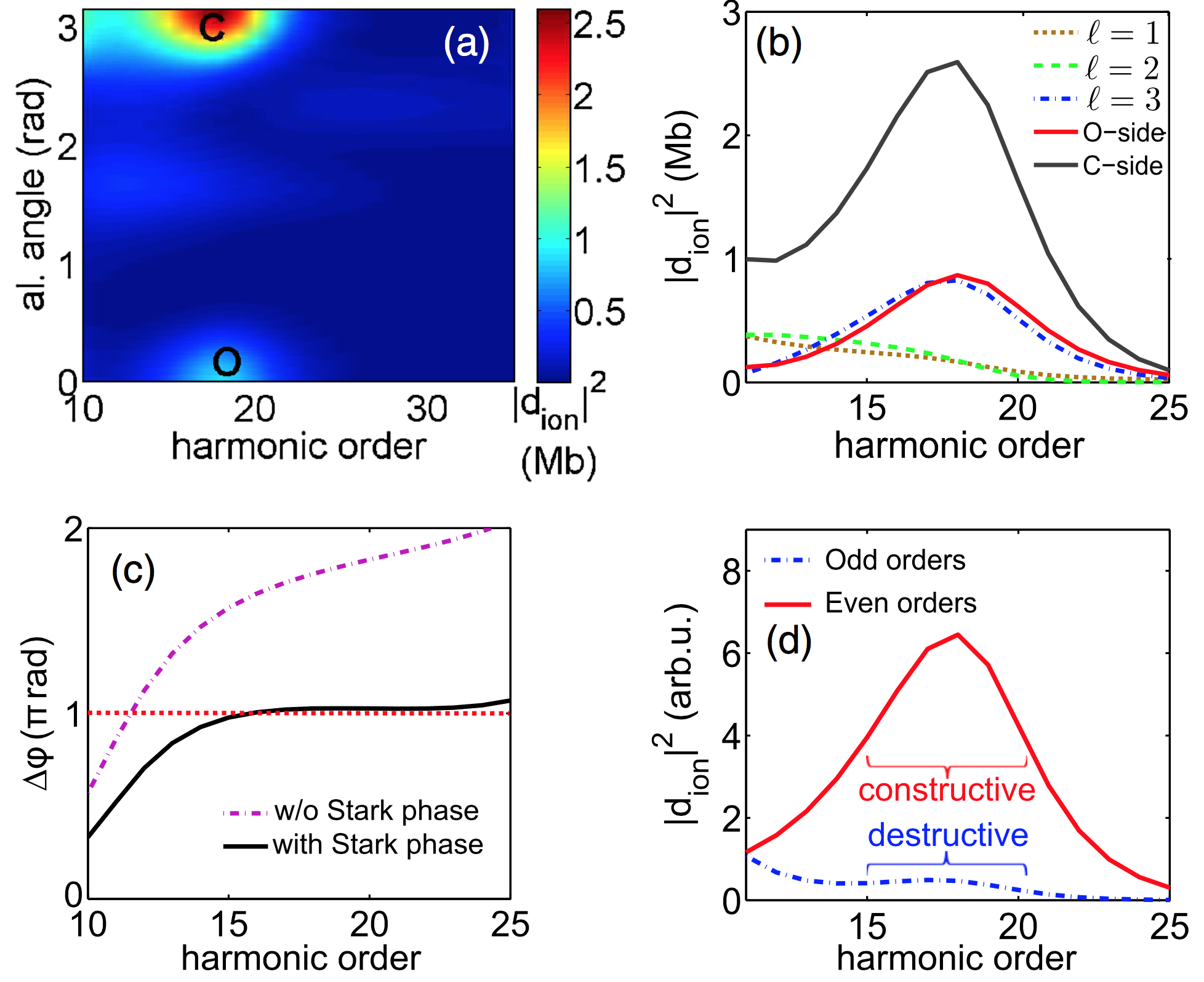
Publications:
[3] P. M. Kraus, A. Rupenyan, and H. J. Wörner
Download "High-Harmonic Spectroscopy of Oriented OCS Molecules: Emission of Even and Odd Harmonics" (PDF, 691 KB)
Phys. Rev. Lett., 109(23), 233903 (2012)
doi: external page 10.1103/PhysRevLett.109.233903
[4] P. M. Kraus, D. Baykusheva, and H. J. Wörner
Download "Two-pulse field-free orientation reveals anisotropy of molecular shape resonance" (PDF, 873 KB)
Phys. Rev. Lett. 113, 023001 (2014)
doi: external page 10.1103/PhysRevLett.113.023001
[5] P. M. Kraus, D. Baykusheva, and H. J. Wörner
Download "Two-pulse orientation dynamics and high-harmonic spectroscopy of strongly oriented molecules" (PDF, 1.8 MB)
J. Phys. B: At. Mol. Opt. Phys. 47, 124030 (2014)
doi: external page 10.1088/0953-4075/47/12/124030external page
High-harmonic spectroscopy features several unique properties for probing photochemical dynamics on the (sub)-femtosecond time scale. First, the high-harmonic emission from excited and unexcited molecules is phase locked, providing interferometric sensitivity to the emission of excited molecules. Second, when combined with the transient-grating technique, both amplitude and phase of high-harmonic emission from the excited molecules can be measured with the unexcited molecules serving as a local oscillator [6]. Third, the technique naturally has a high sensitivity to the transient electronic structure of the molecules arising from the photorecombination step. This property can be exploited to probe the evolution of electronic structure during photochemical dynamics occurring at conical intersections [7]. Recently, the technique has been generalized to larger polyatomic molecules and combined with impulsive alignment techniques [8,9] which will provide molecular-frame measurements of electronic dynamics during chemical reactions.
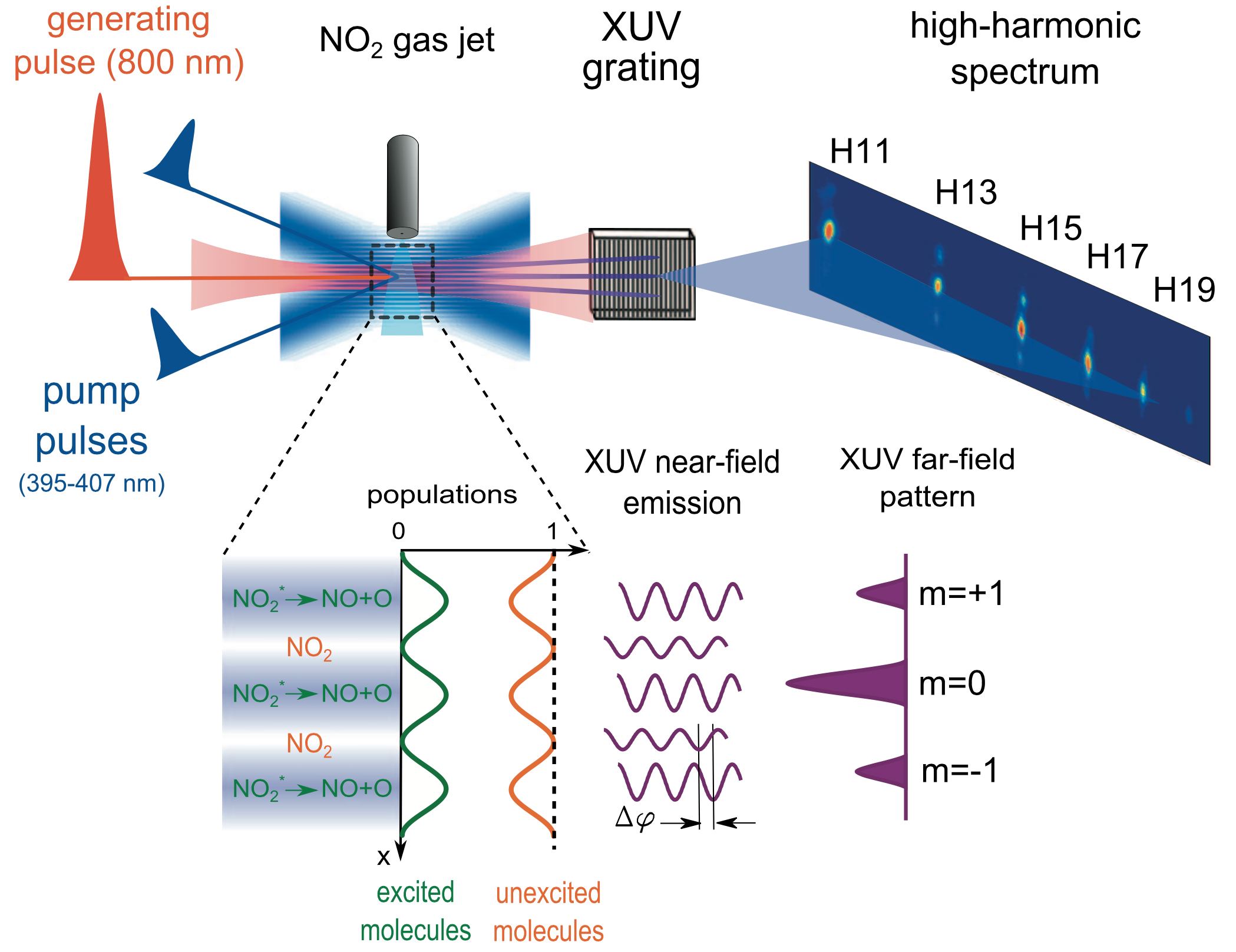
Publications:
[6] H. J. Wörner, J. B. Bertrand, D. V. Kartashov, P. B. Corkum, and D. M. Villeneuve
Download "Following a chemical reaction using high-harmonic interferometry" (PDF, 581 KB)
Nature 466(7306), 604-607 (2010)
doi: external page 10.1038/nature09185
external page Supplementary Information
[7] H. J. Wörner, J. B. Bertrand, B. Fabre, J. Higuet, H. Ruf, A. Dubrouil, S. Patchkovskii, M. Spanner, Y. Mairesse, V. Blanchet, E. Mével, E. Constant, P. B. Corkum, and D. M. Villeneuve
Download "Conical intersection dynamics in NO2 probed by homodyne high-harmonic spectroscopy" (PDF, 1016 KB)
Science, 334(6053), 208-212 (2011)
doi: external page 10.1126/science.1208664
external page Open Access to pdf-file
external page Open Access to full text
external page Supplementary Information
external page Perspective by B. Whitaker: "Shining light on diabolic points", Science, 334(6053), 187-188 (2011)
[8] A. Tehlar and H. J. Wörner
Download "Time resolved high-harmonic spectroscopy of the photodissociation of CH3I and CF3I" (PDF, 959 KB)
Molecular Physics 111(14-15), 2057-2067 (2013)
doi: external page 10.1080/00268976.2013.782439
[9] A. Tehlar, P. M. Kraus, and H. J. Wörner
Download "Probing electronic dynamics during photochemical reactions" (PDF, 627 KB)
Chimia 67(4), 207-212 (2013)
doi: external page 10.2533/chimia.2013.207
High-harmonic generation from coherent superposition states has been widely studied in theory, but has not been observed until 2013 [1]. In addition to the two direct channels (blue arrows in Fig. 1) that correspond to ionization from and recombination to the same quantum state and also occur in incoherent or mixed states, coherence opens two additional cross-channels (red arrows in Fig. 1) that correspond to ionization from one state and recombination to the other state. These channels provide an interferometric sensitivity to weak excitations, turning a 1% excitation into a 55% signal modulation [1]. Applied to molecules, this technique offers a new approach to probing electronic coherence and its dephasing caused by nuclear dynamics [2,3]. The technique also provides access to fine details of the photoionization continua, such as shape resonances and Cooper minima [2]. A detailed theoretical description based on a density-matrix formalism has been developed to describe this technique [3].
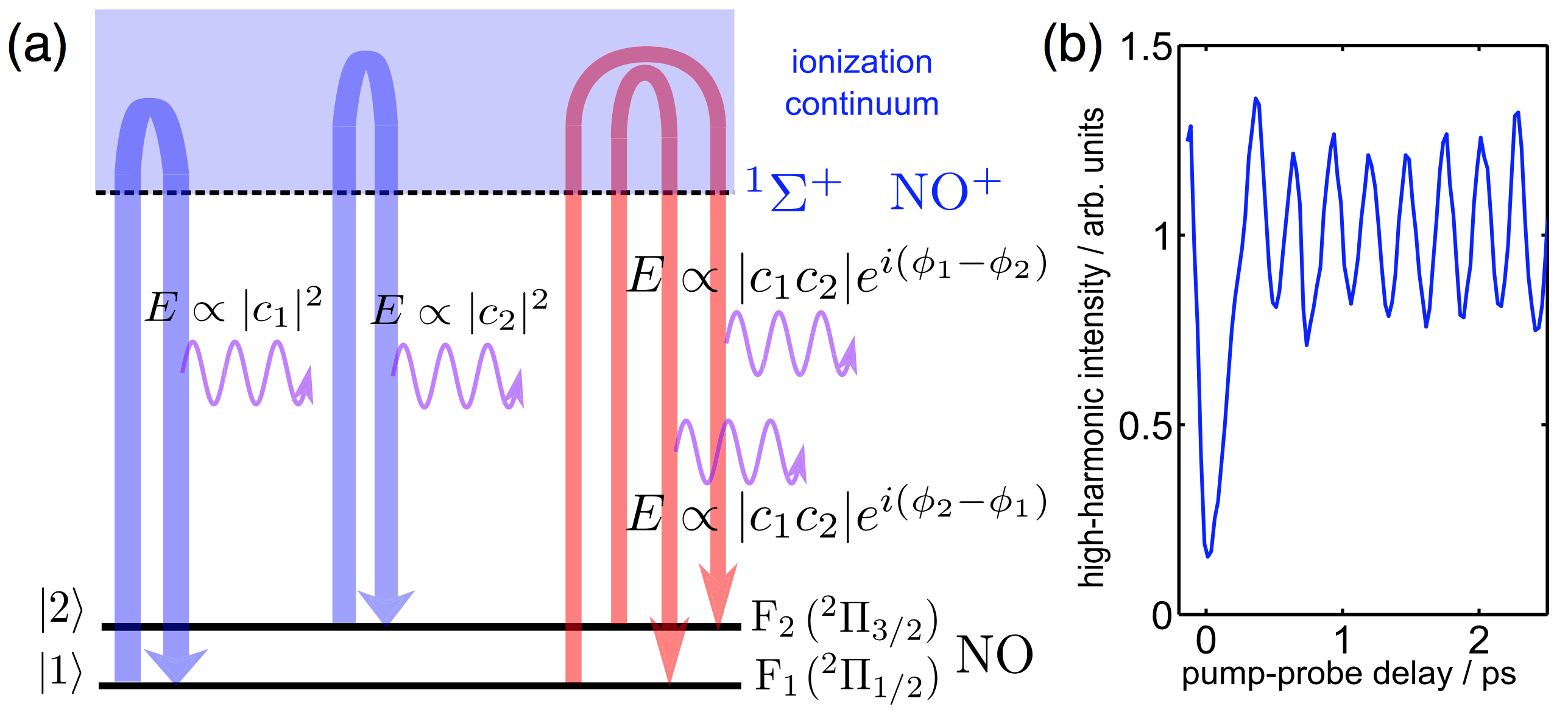
Publications:
[10] P. M. Kraus, S. B. Zhang, A. Gijsbertsen, R. R. Lucchese, N. Rohringer, and H. J. Wörner
Download "High-harmonic probing of electronic coherence in dynamically aligned molecules" (PDF, 908 KB)
Phys. Rev. Lett. 111, 243005 (2013)
doi: external page 10.1103/PhysRevLett.111.243005
[11] D. Baykusheva, P. M. Kraus, S. B. Zhang, N. Rohringer, and H. J. Wörner
Download "The sensitivities of high-harmonic generation and strong-field ionization to coupled electronic and nuclear dynamics" (PDF, 1.2 MB)
Faraday Discuss. 171, 113-132 (2014)
doi: external page 10.1039/C4FD00018H
[12] S. B. Zhang, D. Baykusheva, P. M. Kraus, H. J. Wörner, and N. Rohringer
Download "Theoretical study of molecular electronic and rotational coherences by high-order-harmonic generation" (PDF, 2.5 MB)
Phys. Rev. A 91, 023421 (2015)
doi: external page 10.1103/PhysRevA.91.023421